多肽与蛋白质作为生物体内的活性物质和生命活动的物质基础,在信号传递、能量利用、免疫应答等基础生理过程发挥着至关重要的作用,并与多种疾病的发生密切相关。获得一定数量高纯度的多肽和蛋白质是研究其结构、生物学功能以及开发相关药物的重要前提。天然多肽与蛋白质的来源主要有动植物的组织器官、微生物的次级代谢产物等。目前,自然提取、重组技术和化学合成是多肽与蛋白质的主要获得途径。相较于从天然产物中提取分离和基因重组表达,化学合成能够方便地在多肽与蛋白质的任意位点引入非天然氨基酸或特定类型的翻译后修饰基团,如糖基化、磷酸化、荧光团及光交联反应基团等,极大地促进了多肽与蛋白质在基础医学及生物医药研究领域的应用发展。本综述全面介绍了多肽与蛋白质的各种化学合成研究策略,并讨论了这些策略的基本原理、优缺点及应用价值,旨在为多肽及蛋白质的合成研究提供参考。
人类合成多肽的历史可以追溯到20世纪初,1901年,Fischer等[1]合成了首个无保护的二肽结构(Gly-Gly),并于次年提出了专业术语“peptide”。起初,多肽的合成主要有两种方法:分别是Curtius和Fischer提出的酰基叠氮化物缩合反应[2]和酰氯缩合反应[3]。其中,可移除的暂时性氨基酸保护基的不足是多肽合成的主要制约因素。因此,多种保护基被发明并使用,如Cbz(苄氧羰基)保护基、Boc(叔丁氧羰基)保护基,它们都不同程度上促进了多肽合成的研究进程,许多有名的多肽在这些保护基的帮助下顺利合成,如八肽催产素[4]、β-促肾上腺皮质激素[5]。如今各种氨基酸保护基被发明并使用,已经成为多肽合成研究的重要基石。Merrifield发明的固相多肽合成方法(Solid phase peptide synthesis, SPPS)[6,7]将多肽合成推向新的高度,此方法对该领域的卓越贡献也使他获得了1984年的诺贝尔化学奖。几十年来,随着树脂和缩合剂等的不断发展,固相多肽合成技术发展成熟,已经成为合成多肽的首要选择。多肽合成的快速发展也推动着蛋白质的合成研究。目前蛋白质的化学合成主要分为两部分:多肽片段的合成以及肽片段的连接。如今,各类连接方法发展迅速,允许未经保护的肽链在温和的条件下聚合,使得蛋白质的化学合成更加方便快捷。
虽然多肽和蛋白质的获得有多种途径:自然提取、重组技术和化学合成,但是只有化学合成允许从原子水平上对多肽和蛋白质进行操作,从而实现非天然氨基酸的引入或各种复杂的翻译后修饰过程。化学合成为蛋白质结构的无限变化提供了机会,这对在分子水平上理解蛋白质的结构及功能特性的机制是极其重要的。本综述主要从两个方面开展,在多肽的化学合成这一部分中,以固相合成方法为主,对多肽(特别是困难多肽)的合成策略进行了详细介绍;在蛋白质的化学合成这一部分中,以各种多肽片段的液相连接方法为主,详细介绍了现代蛋白质化学合成的现状。本综述不仅介绍了各种方法的原理、发展及使用步骤,还包括特殊多肽和困难蛋白质的合成方法以及蛋白质的半合成方法。
2 多肽的化学合成
2.1 液相合成法
在固相合成法出现前,液相合成是多肽化学合成的唯一方法,也创造过极大的成就,如八肽催产素[4]、世界上首次人工合成的结晶牛胰岛素[8]。它和大多数的化学反应一样,是在液相中进行氨基酸的缩合连接。尽管固相合成是当今多肽合成的主要方法,但是液相合成因其独特的优势仍在使用,特别是对于一些具有非常规结构的多肽,往往难以通过固相方法合成获得。2016年,Hattori等[9]利用液相合成法成功得到了三骨菌素A(Triostin A),一种具有二硫键的对称双环结构,也是含两个酯键的特殊环肽。2017年,Zhang等[10] 采用“3+2+1”液相缩合策略成功合成了乙酰六肽-3。2014年,Fuse等[11]使用液相方法实现了甘露霉素苷元的全合成。
然而由于反复的缩合、分离纯化等步骤费时费力,这种液相反应并不适合长链多肽或蛋白质的获取,因此固相合成已经成为多肽合成的主流方法。
2.2 固相合成法
自从1963年Merrifield报道了固相多肽合成方法[6,7]以来,多肽及蛋白质[12,13]的化学合成研究进程加速前进。在固相合成过程中,将肽链末端的第一个氨基酸连接到不溶性的树脂上,然后经过反复的偶合和脱保护步骤将氨基酸按顺序连接,肽链延长完毕后再将目标分子从树脂上切割下来。整个过程中除连接在树脂上的肽链外,其他可溶性杂质通过多次的洗涤过滤而除去。这种反复的洗涤过滤代替了液相反应中的纯化步骤,大大提高了肽合成的速率。而且简便的除杂操作允许加入过量的反应试剂促进缩合过程,这也是固相反应优于液相反应的重要一点。
Boc和Fmoc(9-芴基甲氧基羰基)是适用于α-NH2的两种保护基团,分别需要强酸和弱碱两种不同的脱保护条件[14]。尽管利用Boc-SPPS技术获得过许多多肽[15,16],但是由于其需要使用剧毒的氢氟酸进行裂解,而且许多翻译后修饰的结构与氢氟酸并不兼容,这使得条件更加温和的Fmoc-SPPS占据合成肽的天下[17]。目前多篇文献都已详细介绍了Fmoc-SPPS的操作步骤[18,19],基本的过程可以概括为树脂溶胀、连接第一个氨基酸、对未反应的树脂位点用甲醇猝灭、偶联下一个氨基酸、偶联洗涤反复进行至链延长完毕、裂解液处理树脂获得多肽(图1)。固相多肽合成不同于普通的液相反应,无法通过方便的TLC(薄层色谱法)监测反应进程,但是可以通过Kaiser test 监测偶联过程[20],因为未参与偶合的游离氨基(二级胺脯氨酸除外)会被茚三酮染色(蓝色)。
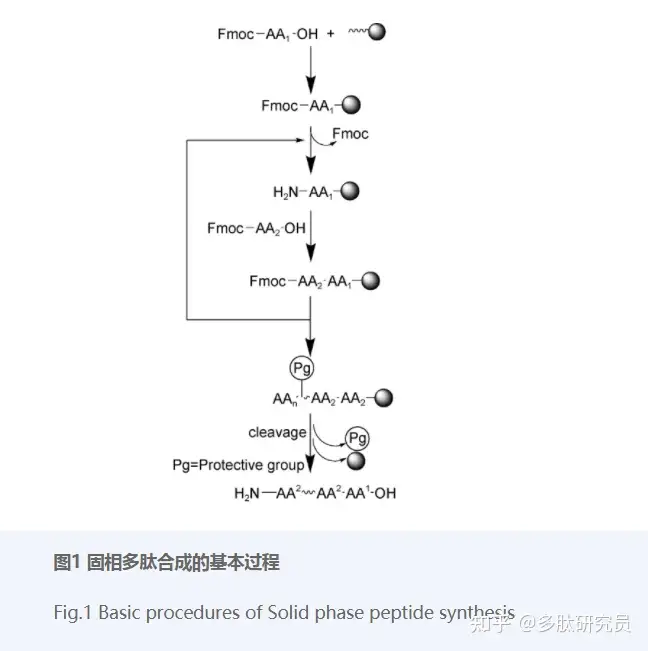
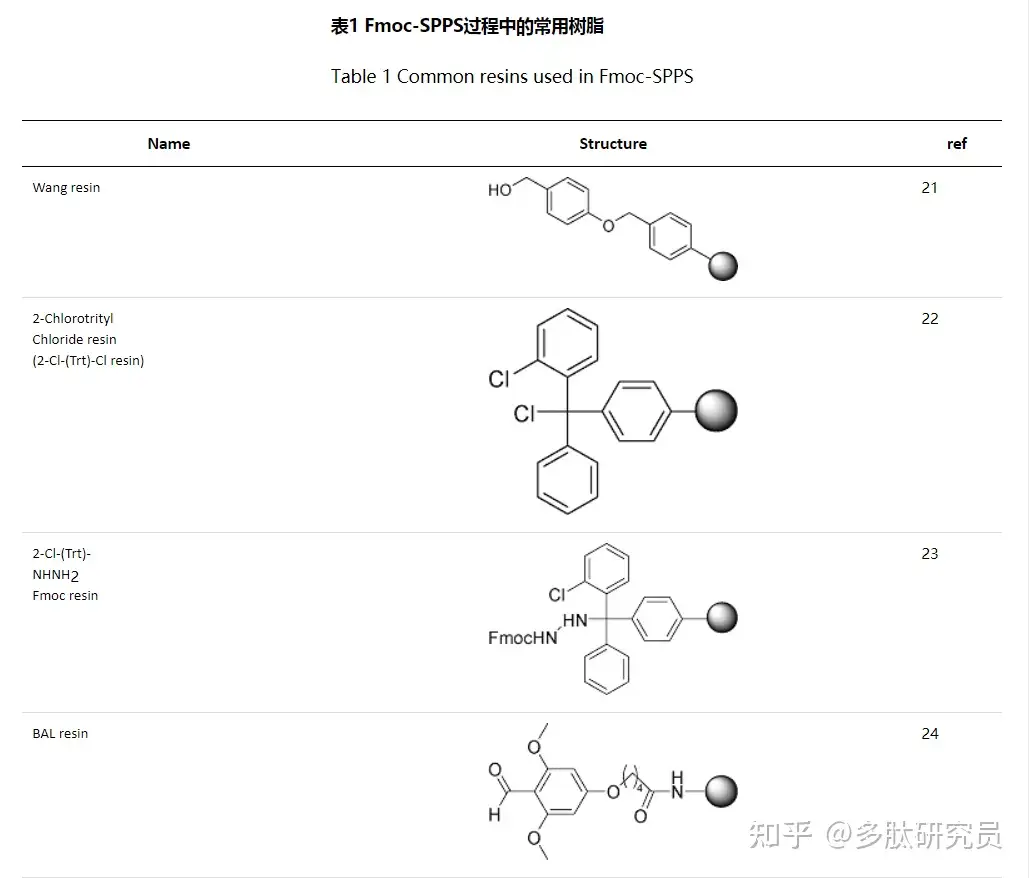
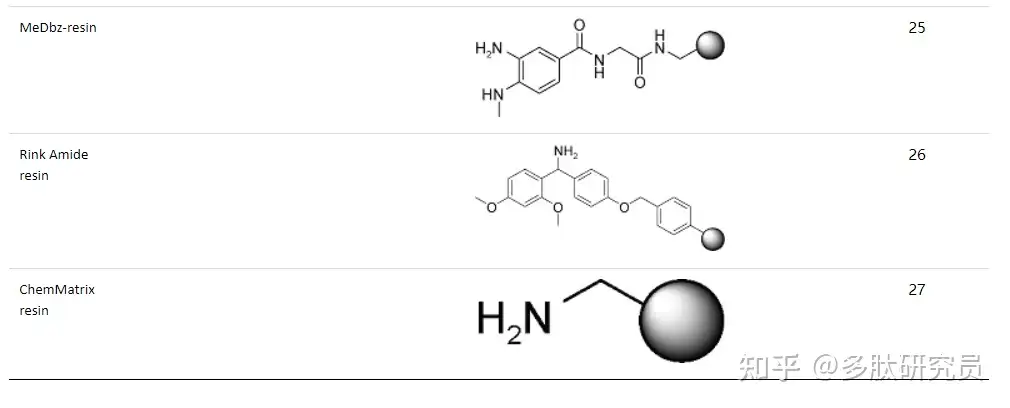
几十年来固相多肽合成技术的核心思想(即其合成步骤)始终未变,但是参与反应的树脂、缩合试剂等不断发展。树脂作为固相状态的来源,由固相载体(solid support,由交联聚合物组成)和连接体(Linker,连接固相载体和目标肽链)组成。它必须满足几个要求:化学稳定性、不溶性、可溶胀性、装载当量合适。固相载体十分重要,因为肽的合成反应发生在其被溶胀的网格间隙内,这种不溶于绝大多数溶剂的结构可以很好地溶胀于DCM(二氯甲烷)和DMF(N,N-二甲基甲酰胺),这也是DCM和DMF用作固相反应试剂的原因。由于肽链的合成方向大多是从C端到N端,所以大多数树脂是锚定肽链C末端第一个氨基酸的羧基。下表1列出了Fmoc-SPPS技术中的常用树脂。
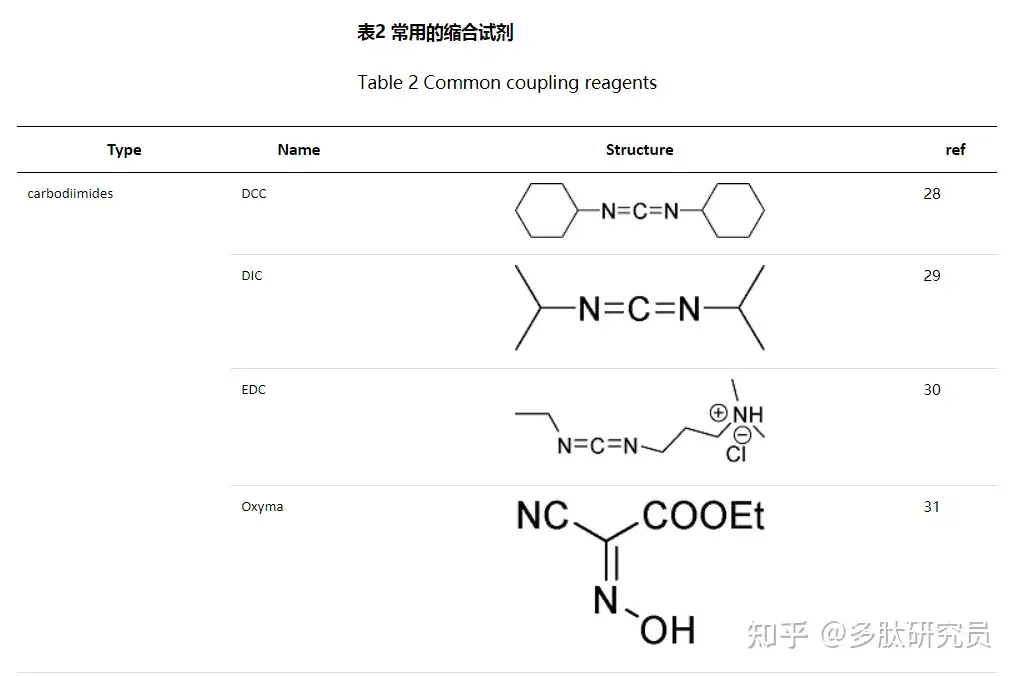
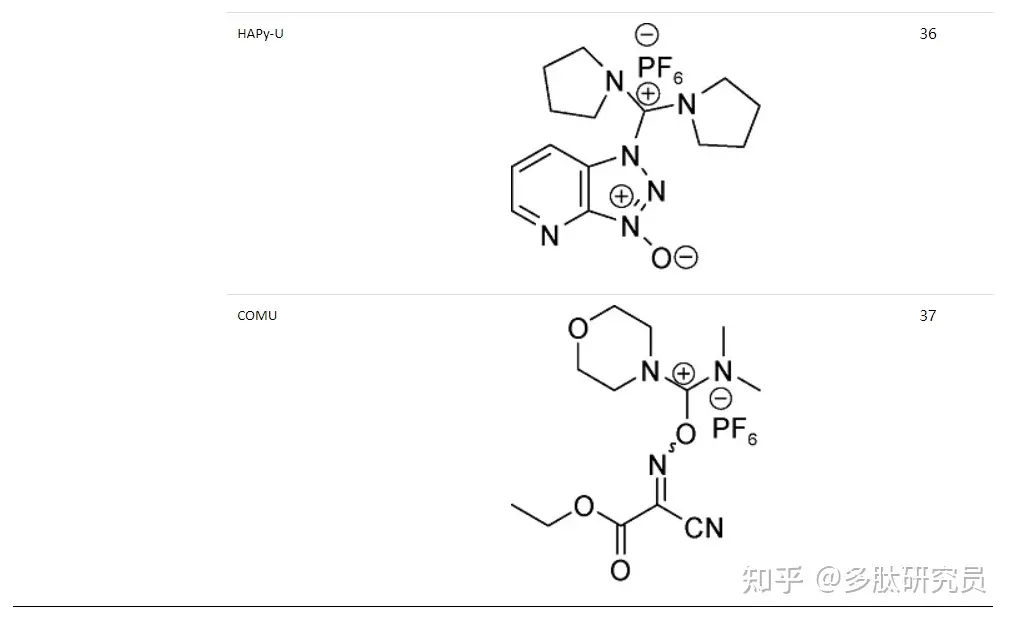
肽骨架是由酰胺键的形成构建起来的,然而酰胺键的形成并非易事。虽然氨基酸的氨基在高pH即碱性条件下是一个很好的亲核基团,但是羧基中的羟基却不是一个好的离去基团,因此羧基必须被激活(CO-X)以允许提供酰基受体接受氨基的亲核进攻,这种激活试剂就是缩合试剂。下表列出了一些常用的缩合剂(表2)。


固相多肽合成法如今已发展得相当成熟,能够允许含50个以内的氨基酸的多肽和一些小型蛋白质顺利合成[38]。为多肽及蛋白质的大量获得做出了伟大的贡献。然而该方法目前依然存在许多缺点,缩合及洗涤过程中对环境不友好的溶剂(DCM和DMF等)和缩合剂的大量使用使得成本过高并引起浪费。因此当合成大规模的短肽时,液相合成法相对更合适。
2.3 困难多肽的合成策略
起初,困难多肽(Difficult Peptides)主要是指有聚集倾向的多肽[39,40],这种聚集倾向在固相合成过程中会影响溶液与树脂上的肽链的接触,从而导致脱保护和缩合反应不完全甚至阻滞。肽链折叠(如β折叠)是引起聚集倾向的主要原因,这种折叠由酰胺氢(—NH)和羰基氧(—C=O)之间所形成的非共价氢键造成(图2)。后来,困难多肽的范围扩大到了所有不易合成的多肽[41],如含疏水性氨基酸导致溶解性差、某个氨基酸缩合困难等等。在Fmoc-SPPS过程中,肽链的聚集使本应该继续反应的N末端闭塞。然而在Boc-SPPS中,用来脱保护的TFA(三氟乙酸)可以有效地破坏肽链的聚集。所以本节中针对困难肽的合成策略主要是应用于Fmoc-SPPS。各种合成策略的适用范围及其特点都总结在表3中。


2.3.1 伪脯氨酸方法
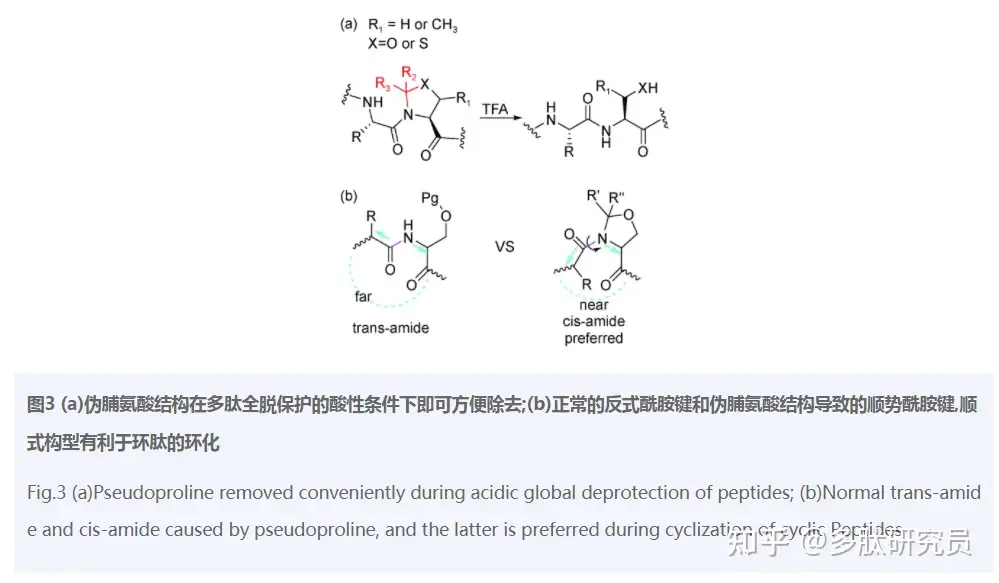
伪脯氨酸方法(Pseudo-Prolines method)由Mutter等于1996年提出[42]。受脯氨酸结构的启发,他们将Thr(苏氨酸)、Ser(丝氨酸)、Cys(半胱氨酸)的α-NH2与侧链的—OH(或—SH)连接成五元环。用这种包含口恶唑烷或噻唑烷五元环结构的伪脯氨酸代替普通氨基酸连接到肽链上,可以阻止氢键的形成从而扰乱肽链的聚积,达到增溶的目的。在肽链延长完成后,伪脯氨酸结构可以在脱保护的TFA条件下恢复到天然的氨基酸残基(图3a)。通常这种伪脯氨酸会以更易缩合的二肽单元的形式连接到树脂肽链上[43⇓~45],如AA-Ser(ψMe,Mepro)。伪脯氨酸方法为复杂多肽的合成开辟了新的前景,并且应用于许多多肽和蛋白的合成。Abedini等[45]于2005年成功合成的人胰岛淀粉样多肽(hAmylin1-37)就是对该方法的有力证明,伪脯氨酸二肽片段的使用不但提高了固相合成的产率,而且方便了纯化过程,使得该困难多肽的合成得到简化。
除了增溶,该策略的应用还有很多。研究发现伪脯氨酸结构对于顺式酰胺键构象的改变可以帮助环肽环合[46](图3b)。利用这一点,Liu等使用伪脯氨酸等方法成功合成了环五肽MZ602和MZ568[47]。从另一角度看,伪脯氨酸结构是对侧链羟基的暂时性保护。2012年,Wang等[48]合成糖肽时,为了防止天冬酰胺副产物的生成,使用伪脯氨酸二肽结构合成肽链,并成功合成了两个关键的糖肽片段:EPO(79-124)和EPO(1-28)。2020年,Zeng等[49]在合成人E-钙粘蛋白N-糖肽的肽链部分时,也使用了伪脯氨酸二肽结构抑制天冬酰胺副产物的生成。总之,该方法作为一种有效的解决思路越来越多地应用于多肽的合成过程。
2.3.2 邻羟基苄基类保护法
与伪脯氨酸方法相似,邻羟基苄基类保护也是通过保护骨架酰胺键从而扰乱氢键的形成。最初的邻羟基苄基保护基是Johnson等提出的N-(2-羟基-4-甲氧基苯)(Hmb),也是首次对骨架酰胺进行保护[50]。一般来说,仲胺较于伯胺需要更加激烈的酰化条件,而且即便是在重复偶合和延长时间的情况下也很难酰化完全。而巧妙的Hmb结构中的邻羟基通过O/N酰基转移起到了助酰化作用(非空间阻碍作用),保证了下一位氨基酸顺利偶合连接。脱Fmoc的N-Hmb结构存在两种互变异构体A和B,构型B利于发生O-酰基化,酯键一旦形成,O/N酰基转移也随之发生(图4a)。2016年,Li等[51]在Hmb结构的帮助下利用STL(丝氨酸/苏氨酸连接)策略成功合成了核蛋白HMGA1a,Hmb结构的引入提高了该蛋白的溶解度,使该合成路线能够获得一个令人接受的多毫克的产量。

Hmb保护基虽然保证了酰化过程的完全,但速度方面却不尽人意。所以基于Hmb一系列改良的保护基结构被报道,目的是通过引入吸电子基团加快酰基化进程,如Hmsb[52]、Hmnb[53](图4b)。同时,强溶胀树脂能力的溶剂也会加速酰基转移,如1,4-二氧六环[52]。
2.3.3 O-酰基化异肽方法
O-酰基化异肽(O-acyl isopeptide)不同于普通的多肽链,它的骨架结构中含有酯键并非单一的酰胺键。这种酯键是Thr或Ser的侧链OH代替α-NH2与下一个氨基酸的COOH缩合而成的。酯键的存在打乱了骨架中酰胺键的连续性,因此阻碍了β-折叠造成的聚集和分子内或分子间氢键的形成。该方法有两个关键的步骤,酯化和酰基O/N的转移(释放天然的酰胺键)[54]。考虑到酯化缩合会引起差向异构化,研究人员将酯连接的二肽单元(如Boc-Ser(Fmoc-Val)-OH)缩合到肽链上而非直接在树脂肽链上酯化[55](图5a),而且现在市售的O-异酰基二肽单元可以方便地获得。

这种O-酰基化修饰不仅有利于困难肽的固相合成,还有助于合成后液相状态的分离纯化。因为O-酰基化异肽可以在酸性条件下稳定存在,因此TFA的裂解液并不会对其照成影响,并可以形成三氟乙酸盐粉末稳定存在(图5b)。所以其酰基O/N的转移可以通过调节pH来诱导发生[56]。除了pH,其他方法也可以用来诱导酰基转移,如光照射。“光触发开关”方法是使用特殊保护基保护Thr或Ser的α-NH2,这种保护基团可以在光照下脱除。2008年,Taniguchi 等使用该方法合成Aβ1-42(一种与阿尔兹海默症相关的肽)[57]。他们将一种香豆素衍生物作为氨基保护基,在多肽脱树脂后保留在肽链上并参与液相纯化过程,由于该保护基的高水溶性,纯化过程得到极大改善。纯化后的O-酰基化异肽在光照下脱去香豆素衍生保护基,并在中性缓冲液中进行酰基迁移得到目标肽(图5c)。2021年,Hojo等[58]通过异肽结构增加蛋白溶解度的方法,成功合成了由177个氨基酸残基组成的膜蛋白Caveolin-1。
2.3.4 聚乙二醇化方法
聚乙二醇(PEG)是一种末端连羟基的线性或支化聚醚,因此可以利用这种结构带来的高溶解性对生物分子进行修饰——聚乙二醇化(Pegylation)。同样,将聚乙二醇结构单元引入到肽链将极大地增加多肽链的水溶性,不仅利于SPPS过程中肽链的偶合延长,更重要的是利于后期液相纯化。所以,将聚乙二醇成功的接入多肽链是关键。1994年,Lu等[59]将聚乙二醇结构引入肽序列,使用SPPS来合成聚乙二醇化的多肽。他们使用不同的连接方法将聚乙二醇引入到不同的位点:NH2末端、侧链处(赖氨酸/天冬氨酸)、COOH末端。比如,对于引入到侧链处的两种方法:将提前制备好的已连接PEG的氨基酸(图6)偶合连接到肽链;或将肽链按标准程序延长完成后,再将PEG加入到树脂多肽上。PEG作为一种优良的增溶剂,是多肽优化策略的重要一员,其对天然多肽或蛋白的修饰,能够有效改善结构并获得更好的生物学功能。如,2021年,Yang等[60]在某段胰高血糖素的24位引入半胱氨酸残基以实现多肽的聚乙二醇化;2020年,Wang等[61]借助谷氨酸将PEG6连接到目标肽的N末端以实现肽稳定性和靶向性的增强。

2.4 其他合成策略
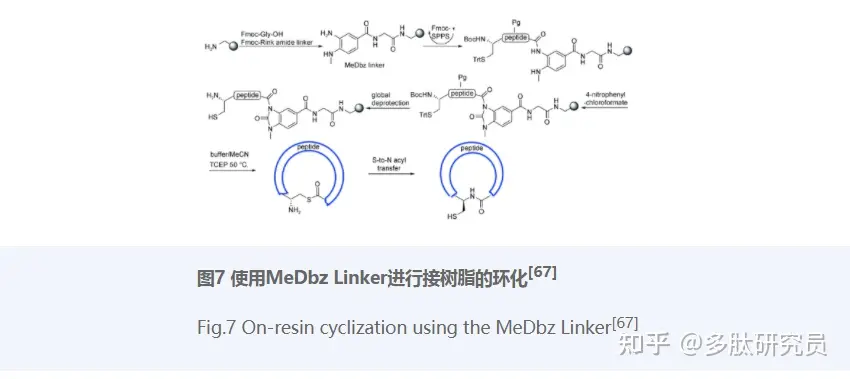
环肽由于其特殊的结构和药理学特征,如强亲和力、低毒性等,为药物设计提供了更广泛的空间,并成为近年来的研究热点[62]。线性肽在固相合成技术的帮助下已经可以轻松合成,然而线性肽的环合却为环肽的获得带来了不少挑战。头尾缩合(或头与侧链、侧链与侧链、侧链与尾)的方式进行环合是最直接的方式,然而这种直接缩合的方式存在的副反应是难以避免的,如分子间的缩合、C末端手性结构的破坏[63]等。自然化学连接(NCL)的发展为环肽的环合提供了新方法,该方法不仅有效降低了C末端的差向异构化,还允许线性肽在全脱保护的状态下环合。除了NCL[64],STL[65]、KAHA(α-酮酸/羟胺)ligation等化学选择性的方法都可应用于环肽合成[66],这些连接技术在下文中有详细的介绍。接树脂的环化(On-resin cyclization)也是环肽合成的一个重要方法,其重要的优势是能方便地去除溶剂和过量的反应试剂。2018年,Gless等[67]报道了一种利用邻氨基(甲基)-苯胺(MeDbz)作为连接体在树脂上环化合成环肽的有效方法(图7)。
糖基化是一种常见而且复杂的翻译后修饰,天然的糖基化过程分为两种:O-糖苷,聚糖与丝氨酸、苏氨酸或络氨酸的羟基形成α或β连接;N-糖苷,聚糖与天冬氨酸的侧链酰胺形成β连接。这种糖基化结构的复杂性给它的合成带来许多挑战,其通常无法通过生物表达的方式获得[68],而化学方法和化学酶的干预却对该特殊多肽的合成有效。2022年,Ma等[69]报道了一种液相化学结合化学酶合成复杂糖肽的方法,多个糖肽的简便合成证明了该方法的广泛适用性。2020年,Zeng等[49]化学合成了人类E-钙粘蛋白N-糖肽,其中天冬酰胺位点的糖苷化过程通过固相合成实现并具有化学选择性。
3 蛋白质的化学合成
各种机智的方法为多肽的化学合成提供了无限可能,液相和固相合成是肽链缩合延长的两种思路,各种酰胺骨架的保护策略有效改善了肽链合成中的聚集和难溶性等问题,并且这些方法也已广泛应用于环肽、糖肽等特殊多肽的合成。然而多肽的这些常用方法只能合成较短的肽链,很难合成大分子的蛋白质。蛋白质不仅分子量(50~300个氨基酸残基或者更多)要比多肽大,线性多肽链的进一步折叠等也使它的结构更加复杂。因此蛋白质不能简单地理解为更大的多肽,它的化学合成也更加复杂,主要思路还是利用固相合成获得几个多肽片段,然后再在液相反应中将这些肽段进行连接,因此称为固相/液相组合合成法(combined solution/solid-phase synthesis)。固相多肽合成法在上文已进行了详细阐述,下文将总结肽段化学连接的主要方法,表4总结了各种多肽的连接方法。

3.1 NCL
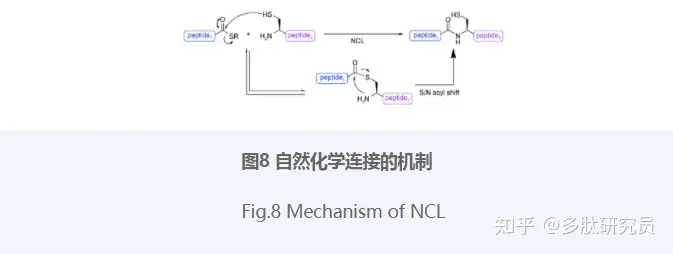
自然化学连接(Native chemical ligation, NCL)[70]的提出推动了多肽连接化学的发展,它允许溶解性更好的脱保护肽段在一个普遍的条件下进行连接,这种方便的连接为蛋白质的合成做出了极大的贡献。自然化学连接形成天然酰胺键的过程与固相合成中的差别很多,它是利用N末端Cys的侧链-SH(软碱)进攻C末端硫酯(软酸)的酰基碳形成一个暂时的酰硫键,然后再通过分子内S/N酰基转移形成天然的酰胺键(图8)。该方法涉及两个肽段,C末端硫酯多肽片段和N末端为半胱氨酸的多肽片段。研究表明芳基硫酯是自然化学连接中高效的参与者,许多外源硫酯激活剂被开发,如MPAA(4-巯基苯乙酸)[71]。因此许多高效的方法用来合成惰性的肽前体,从而能够在NCL条件下转化成活性芳基硫酯参与连接反应。
3.1.1 多肽酰肼结构
多肽酰肼作为优良的前体结构,不但能够在Fmoc-SPPS条件下方便生成,而且很容易被激活成硫酯结构参与NCL。2011年,Fang等[72]报道了这种以酰肼结构为前体的自然化学连接反应(图9a)。首先在自制的酰肼类王树脂上,利用Fmoc-SPPS合成肽酰肼,然后在含亚硝酸钠的缓冲液中氧化成酰基叠氮化物。再加入硫醇将其硫解成硫酯,这样最初的酰肼结构便被激活,进而可以与另一片段的半胱氨酸位点连接。该方法中肼结构的氧化、硫解和肽段连接是让人喜欢的“一锅反应”。遗憾的是,当多肽片段1的C末端氨基酸为Asp(天冬氨酸)/Asn(天冬酰胺)和Gln(谷氨酰胺)时,该方法并不适用,原因是这三种氨基酸脱保护的侧链会与末端的肼结构成环,阻止C末端硫酯的形成。
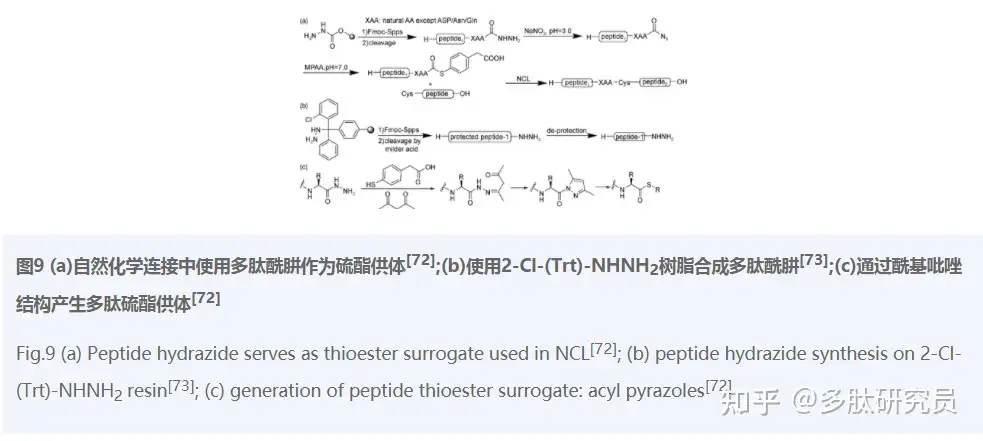
为了克服Asp、Asn和Gln发生副反应而成环的问题,Tian等[73]用一种新的树脂(2-Cl-(Trt)-NHNH2 resin)合成多肽酰肼(图9b)。与之前的方法不同的是,该树脂可以在温和的酸性条件下裂解获得侧链保护的多肽,然后再进行脱保护,从而避免了C末端的第一个氨基酸侧链和“肼”结构发生成环副反应。为了延长这种树脂的储存期从而减少副反应,研究人员又引入Fmoc保护基合成了2-Cl-(Trt)-NHNH2 Fmoc树脂[23]。
酰肼转化为硫酯是该方法中的关键步骤,上文所讲是通过NaNO2的氧化实现的,然而某些基团不能适应含NaNO2的氧化条件,比如N末端噻唑或者其他对氧化还原敏感的基团,所以更加优良温和的肽酰肼激动条件需要产生。2018年,Flood等[74]针对该限制报道了一种温和的硫酯合成条件。使用乙酰丙酮把肽酰肼激活成酰基吡唑中间体(一种高效的酰化剂),再进一步硫解成多肽硫酯参与自然连接反应(图9c)。同样,该固相合成后的一系列连接过程也是通过“一锅反应”进行的,不需要任何中间纯化和分离操作。
除了可以利用SPPS合成肽酰肼结构,表达蛋白和酶催化[23]都可以形成此类结构从而应用于NCL。
3.1.2 隐形肽硫酯
Terrier等[75]发明了一种方便合成的隐形硫酯装置N-Hnb-Cys(StBu),可以在中性pH条件下高效快速地获得NCL所需的肽硫酯片段,且NCL在该方法中完全可行。较其他方法相比,本方法简便高效,两个多肽片段通过固相合成后,只需一步反应就可实现连接得到目标多肽。如图10,SH的保护基StBu(叔丁基巯基)在NCL条件下即可快速脱除,且硫醇暴露后就会触发下一步的反应;分子内的N/S酰基转换是借助Hnb保护基高效合成肽硫酯结构。该方法已经成功地应用于合成两种富有Cys的多肽序列:MT7和Cg-BigDef1。
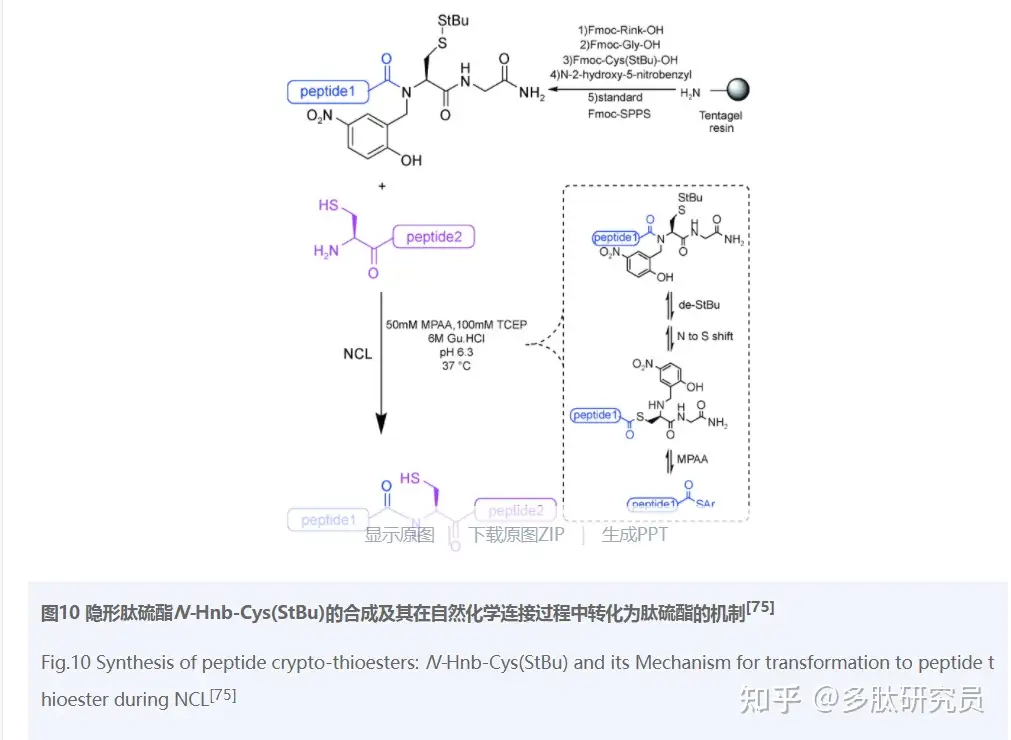
隐形肽硫酯是一种优良的硫酯代替物,但是当Ile(异亮氨酸)、Val(缬氨酸)和Thr或Pro作为隐形肽硫酯的C末端氨基酸时,在Fmoc-SPPS条件下这些特殊的氨基酸与N-Hnb-Cys结构偶联产率极低。Lelievre等的研究表明,较慢的O/N酰基转移过程是造成这些氨基酸偶联困难的原因[76]。因此,他们对这步偶联过程进行进一步优化,当第一个氨基酸(如Pro)连接后,用盐酸羟胺和咪唑的混合溶液条件反应消除不需要的酯化产物,增加所需的仲胺酰化。同样也对NCL过程进行了条件优化,pH值变为5.8,温度升高到50℃。
3.1.3 硫醇化氨基酸
NCL的提出对于蛋白质合成的意义重大,但是该方法只能限制于含Cys残基的肽链,况且半胱氨酸在天然蛋白质中的含量极低(1.2%),因此极大限制了它的应用。为了扩大NCL的应用范围,各种方案被进一步提出。其中最重要的一个解决方案就是使用Cys的类似物进行连接,最后再通过脱硫除去巯基获得想要的氨基酸侧链。起初脱硫是使用雷尼镍(Raney nickel)或者钯/氧化铝(Pd/Al2O3)[77],然而这两种条件都存在一定的局限性。雷尼镍会引起仲醇差向异构化以及硫醇、硫酯和硫醚的还原,作为N末端半胱氨酸残基保护基的噻唑烷结构(Thz)在钯/氧化铝的条件下不稳定[78]。所以,温和的脱硫条件亟需提出。2007年,Danishefsky等[78]通过自由基介导还原实现了半胱氨酸脱硫转化为丙氨酸,这种温和的条件(TCEP,三(2-羧乙基)膦)适合于复杂多肽和糖肽的合成。2016年,Pasunooti等[79]利用此脱硫条件将硫醇化的Ile转化为天然的氨基酸残基(图11a),类似的脱硫反应在蛋白质合成领域应用广泛[80,81]。

越来越多的硫醇化氨基酸也已经被合成: γ-thiol Ile[79]、β-thiol Lys[82]、γ-thiol Pro[83](图11b)等。2020年,Yin等[84]报道了一种高效合成β硫代(Cys类似物)/硒代氨基酸的光催化不对称反应,实现了β-硫代/硒代氨基酸的克级制备。总之,高效的脱硫反应和这些硫醇化的氨基酸变构体的发展为NCL的应用开创了新的天地。
3.2 DSL
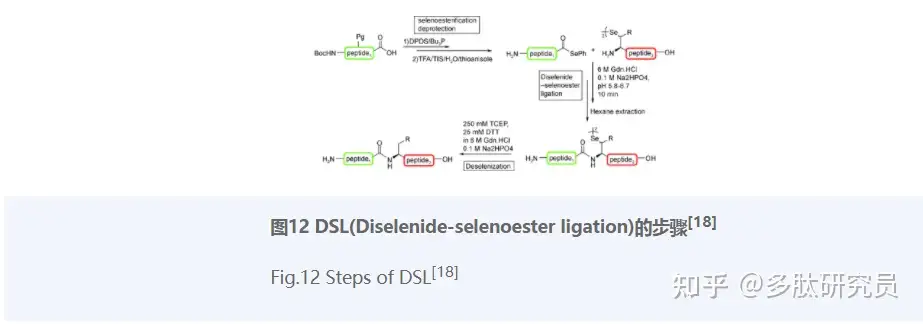
NCL的固有局限性促进了替代性化学选择性连接技术的发展,如DSL(Diselenide-selenoester ligation)。DSL与NCL十分相似,其是在一条C端芳基硒酯肽片段和另一N端硒代半胱氨酸肽片段之间进行连接,然后再通过脱硒获得目标多肽(图12)。该方法可用于蛋白质的快速合成,包括不能使用或不能有效使用其他肽连接方法的情况。2019年,Kulkarni等[18]使用该方法成功合成了3条多肽蛋白:脂联素(19-107)、血红素1和血红素2。2022年,Liczner等[85]报道了一种使用DSL和去硒化策略合成肽-寡核苷酸结合物(与肽结合的修饰寡核苷酸)的有效方法。
3.3 STL/CPL
STL(Ser/Thr ligation)[86]与NCL不同,其是由水杨醛酯作为酰基供体,与N末端为Ser或Thr的多肽片段进行连接。首先,水杨醛结构中的醛基与Ser或Thr的羟基和氨基反应形成五元噻唑环,两条肽片段因此连接起来。其次,水杨醛拥有和Hub类似的邻羟基结构,通过六元环过渡态进行O/N酰基转移形成天然的酰胺键(图13)。更为巧妙的是连接完成后,通过合适的酸化条件就可获得目标肽链,水杨醛就像是一个锁,两条肽链连接后即可轻松移除。STL满足多肽连接所需要的化学选择性,因为醛基与氨基形成亚胺的过程是可逆的,侧链上的氨基位点形成亚胺后由于缺少羟基基团阻碍了进一步的反应,从而实现化学选择性。在适用范围方面,20种天然氨基酸除了Asp、Glu(谷氨酸)和Lys(赖氨酸)都可以作为C末端参与该连接反应[87]。此外,该方法在合适比例的吡啶醋酸混合液中便可连接,不需要复杂的操作或设备,因此被广泛应用于多肽和蛋白质的合成[88,89]。

类似于NCL中的肽硫酯,肽水杨醛酯(SAL ester)也是STL的必需,它的制备也有许多方法。直接缩合(Direct coupling)是将全保护的C端羧酸多肽与二甲氧基水杨醛(二甲氧基保护基可在酸性条件下恢复醛基)在PyBop(六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷)和DIEA(N,N-二异丙基乙胺)的条件下直接缩合成酯。然而这种直接缩合只能应用于C末端为Gly(甘氨酸)和Pro的情况下[90],因为这种缩合条件会造成末端氨基酸的差向异构化。n+1策略[91]却成功避免了差异化问题,而且允许STL从N端到C端进行连接(N to C ligation)。如图14a,n指的是经过Fmoc-SPPS合成的全保护肽段,1指的是醛基被缩氨基脲保护的单氨基酸水杨醛酯,n和1在合适的条件下缩合连接,再经TFA和pyruvic acid(丙酮酸)分别脱除侧链保护基和醛基保护基。

某些空间要求较高的氨基酸作为肽末端氨基酸时,其连接往往受到阻碍(图14b)[92],如Pro、Val、Ile(异亮氨酸)、Thr,它们在自然界的蛋白质中的含量高达22%,所以更好的连接方案需要被提出来解决这些问题。2020年,Tan等[93]基于STL研究出一种具有化学立体选择性的多肽片段的连接方法CPL(Cys/Pen ligation)(图14c)。整个连接过程不需要巯基作为亲核基团进攻末端酰基碳,因此不会受C末端氨基酸高空间位阻的影响。在Tan等的报道中,这种方法的连接率高达95%,且在连接过程中连接位点的两个氨基酸无差向异构作用。CPL作为STL的补充,使得该类连接方法的应用范围更加广泛。
3.4 KAHA ligation
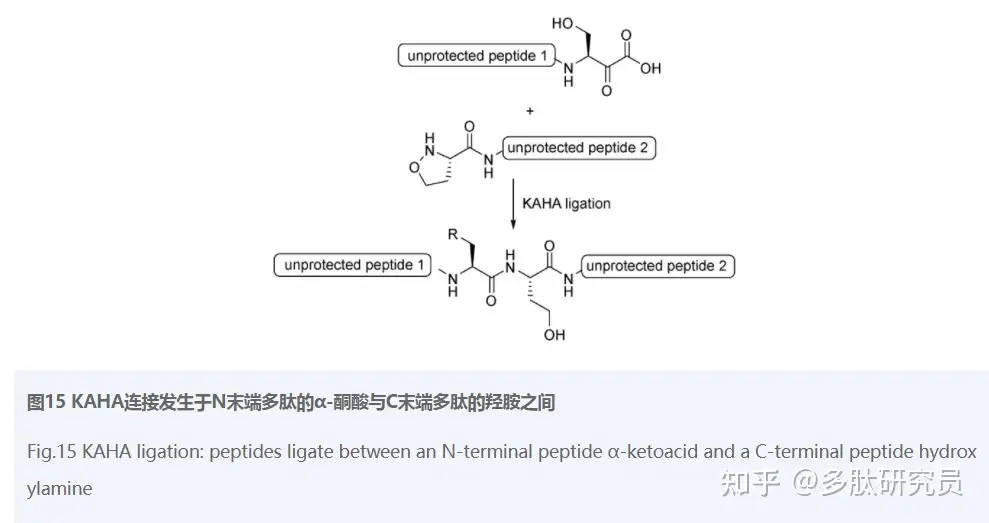
2006年,Bode等[94]发明并报道了一种新的具有化学选择性的连接方法KAHA (α-Ketoacid hydroxylamine) ligation(图15)。与其他的连接方法不同,该法是通过α-酮酸(KA)与羟胺(HA)反应成酰胺键进行连接,研究表明N末端肽段可以有两种变体[95]:O未被取代的羟胺和O取代羟胺,其中五元环状羟胺最常用[96]。除了具有良好的化学选择性、条件温和无需催化剂等普遍的优点之外,KAHA连接最吸引人的另一大优点就是副产物只有二氧化碳和水。多种蛋白质的成功合成都证实了该方法的实用性[97,98],更多有关KAHA 连接方法合成蛋白质的细节囊括在这篇综述中[99]。
3.5 困难蛋白质的合成策略
3.5.1 增溶标签
占人类基因组编码蛋白质约20%~30%的膜蛋白(membrane proteins)[100]对生物学、药学等领域的重要性不言而喻,它是细胞发挥作用的重要媒介,有研究表明高达50%的药物靶点与膜蛋白有关[101]。膜蛋白作为困难蛋白质的一种,它的强疏水性一直是化学合成过程中的难题。把一些额外的溶解性好的氨基酸引入到目标肽中,从而实现具有所需特性(如增溶性)的融合结构体,这种额外的基团被称为“标签(tag)”。这种可移除的暂时性标签是解决膜蛋白难溶性的重要手段。当这种增溶标签连接到侧链上时,又可以起到骨架修饰的作用,因此在增溶的同时又可以在SPPS过程中阻止支链肽的聚集。常见的增溶标签有聚精氨酸(Poly-arginine tag)、聚赖氨酸(Poly-lysine tag)[102](图16a)等,这种由氨基酸组成的基团很容易连接到目标肽中,因此广泛应用于膜蛋白的合成。

2016年,Zheng等[103]介绍了一种利用增溶标签合成膜蛋白的通用方法。他们将聚精氨酸增溶标签通过Hmnb结构接入多肽组成了可移除式骨架修饰基团(RBM-Tag),有效解决了蛋白质的聚集性和难溶性问题,并成功合成了双/四跨膜蛋白质(离子通道蛋白HCV p7和转运蛋白EmrE)(图16b)。最近Liu等[90]也报道了一种借助增溶标签合成膜蛋白的方法,并提出了一种可被还原的增溶标签(reducible solubilizing tag,RST)。RST是一种易安装又易拆卸的方便工具,它与Cys/Ala(丙氨酸)侧链或水杨醛酯通过二硫键连接(硫醇化Ala需再次脱硫),连接完成后再用TCEP(三(2-氯乙基)磷酸酯)或脱硫环境(TCEP和 tBuSH)移除(图16c)。最后,他们使用该策略成功合成了蛋白质2B4(一种由CD244基因编码的人类蛋白质)和膜蛋白FCER1G。
3.5.2 蛋白质的半合成
蛋白质的半合成(Protein Semisynthesis)是介于表达蛋白和化学蛋白之间的一种高效方法,其中至少有一个结构块是由表达而获得,剩下的部分由化学合成。半合成结合了表达蛋白的简便性和低成本以及化学蛋白对特殊位点的可修饰性等优点,此外也可以很好地避免全合成无法使蛋白质折叠的问题[104]。半合成弥补了全化学合成(total chemical synthesis)在大蛋白获得方面的不足,统计显示全合成获得的蛋白质的大小平均在90个氨基酸残基左右,而半合成能获得200个氨基酸残基以上的大蛋白[105]。尽管半合成对于大蛋白的获得具有很多优势,但是还存在许多不足。比如由于缺少合适的半胱氨酸硫醇保护基,使得脱硫反应无法在天然半胱氨酸残基的存在下选择性进行,因而阻碍了表达蛋白连接及随后的脱硫反应。2020年Wang等[80]报道了一种由光控释放的半胱氨酸硫醇保护策略,并成功应用于磷酸化蛋白胱抑素S(CST4)的半合成。
蛋白质的半合成有很多策略,包括表达蛋白的化学连接(Expressed Protein Ligation, EPL)[106,107]、酶催化蛋白连接[108]、酶催化表达蛋白连接(enzyme-catalyzed EPL)[109]等。在各种策略的帮助下,多种特殊的蛋白质被成功合成,包括各种翻译后修饰蛋白质[110]、环肽[111]、组蛋白等等[109]。蛋白质翻译后修饰(posttranslational modifications, PTMs)包括糖基化(glycosylation)[112,113]、磷酸化(Phosphorylation)和乙酰化(Acetylation)等等,化学合成(包括半合成和全合成)是获得该类特殊蛋白质的重要工具,并有助于此类翻译后修饰机制的研究。而当全合成无法使用时,半合成就显示出它的独特优势。2017年,Tan等[23]在合成磷酸化蛋白质时发现某段N端肽链无法通过固相合成获得,进而转用半合成方法(转肽酶催化的蛋白连接)。如图17,通过生物表达合成N端肽链后再在酶的催化下形成酰肼结构,进而与固相合成的肽段进行连接获得目标蛋白质。

4 结论与展望
多肽及蛋白质的化学合成是当代合成化学与化学生物学领域的重要研究热点,一直备受学术界和工业界的高度关注。本综述从多肽合成与蛋白合成两个方面,对当前的化学合成策略及其应用进行了总结和展望。SPPS作为多肽合成的重要手段,已在相关领域得到广泛应用,新型树脂与缩合剂的发展使其日渐完善。针对合成困难的复杂多肽,多种有效的策略相继被提出,如伪脯氨酸方法、邻羟基苄基类保护法、O-酰基化异肽方法、聚乙二醇化方法等,使得“困难”多肽的固相合成顺利可行。此外,针对具有特殊结构的环肽与糖肽,各种有效的合成方法不断被改良创新。比如,多肽骨架酰胺键保护基伪脯氨酸不仅可以通过破坏氢键的形成减少多肽链的聚集达到增溶目的,还能造成肽链空间结构的改变,从而促进环肽的环合过程。随着各类高效连接技术的发展,例如NCL、DSL、STL、KAHA ligation等,多数中小规模的蛋白质合成可以通过固相和连接方法的结合顺利完成,而一些含复杂序列或大规模的蛋白质则需要采取专门的策略或转用半合成的方法。
多肽及蛋白质的化学合成不仅具有重要的理论意义,还在临床检测以及药物研发等领域具有广泛的应用价值。通过化学合成方法在特定的位点引入修饰基团,能够有效地改善多肽及蛋白类分子的理化性质,提高生物活性与稳定性,促进成药。多肽合成技术与组合化学结合,能合成出含有大量化合物的组合肽库,用于筛选具有特定药理活性的先导化合物。尽管多肽及蛋白质的化学合成领域已经取得了长足的发展与斐然的成果,但诸多固有的局限性依然存在。对于“困难多肽”的合成来说,多种策略都围绕着对其进行某些化学修饰以达到更好的溶解性,从而有利于对其的各种操作及纯化等过程。然而这些策略适用范围窄,而且程序复杂费时,所以低成本且能适用于“困难多肽”的通用方法还需要进一步研发。要想通过固相多肽合成法获得一定量高纯度的含30个氨基酸残基以内的普通多肽往往需花费数周的时间,而且很难达到克级别以上。近年来,流动化学(flow chemistry)的快速发展为化学合成多肽和蛋白提供了新的方法,这种借助自动化快速流动仪器进行合成的方法可以允许多肽链在数小时内延长完毕[114],大大提高了多肽和蛋白的合成效率。多肽合成过程中不可避免地需要使用过量缩合剂和对环境不友好的洗涤溶剂(DCM、DMF),所以开发更加高效的缩合方式和绿色环保的溶剂是多肽和蛋白化学合成领域需要解决的问题。目前各种多肽连接方法也仅限于实验室研究,绝大多数并未广泛应用于实际生产中,更加高效的化学连接新方法依然亟需。总而言之,现有的合成策略还无法满足人们对多肽及蛋白质深入探索的需求,更加成熟、普适、简便的方法还需进一步解锁,从而继续推进多肽及蛋白质在分子生物学与药理学研究中的进程。
